
SINDROMI DEL PRIMO E SECONDO ARCO BRANCHIOALE:
sindrome di Goldehar e Micorsomia emifacciale; sindrome di Franceschetti (Treacher-Collins); sindrome di Nager - sindrome di Miller
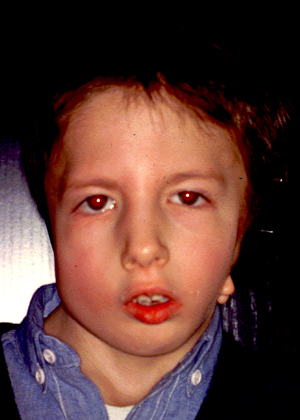
Le Sindromi del I e II arco branchiale sono malformazioni del volto che interessano soprattutto la parte laterale della faccia, si presentano in quadri sindromici monolaterali, come la Sindrome Otomandibolare o Microsomia Emifacciale e la Sindrome di Goldenhar, o bilaterali, come la Sindrome di Franceschetti o Treacher – Collins, la Sindrome di Nager.
Possono interessare, la catena ossiculare acustica, il padiglione auricolare, il nervo facciale che muove il volto, i muscoli masticatori, la mandibola e l’articolazione tra la mandibola e il cranio. Più raramente troviamo Macrostomia (apertura della bocca allungata), deformità delle palpebre, dell’orbita, del bulbo oculare e del cranio.
Dal punto di vista funzionale le forme monolaterali sono spesso asintomatiche, il trattamento inizia dopo i 7 – 8 anni ed è rivolto alla correzione dei difetti delle ossa e dei tessuti molli al fine di migliorare l’aspetto asimmetrico del volto, qualora esistano esigenze psicolodiche da parte del bambino. Il padiglione auricolare può essere ricostruito o corretto da un chirurgo esperto verso i 10 anni.
Anche le forme bilaterali lievi ricalcano lo stesso andamento delle monolaterali mentre in quelle gravi si possono avere alterazioni della funzione respiratoria già dalla nascita che richiedono trattamenti di allungamento mandibolare al fine di evitare la tracheotomia.
L’udito può essere in parte deficitario sia nelle forme bilaterali che monolaterali con una compromissione che è sempre parziale anche nei casi dove i meati acustici (canali dell’orecchio) sono assenti e viene trattato con apparecchi acustici di diverso tipo.
La masticazione e la deglutizione sono sempre presenti quindi sono rarissimi i pazienti che hanno problemi ad alimentarsi fatta eccezione per quei casi dove sono bloccate le articolazioni della mandibola (ad esempio a volte nelle sindromi di Nager). Le anchilosi mandibolari possono essere sbloccate chirurgicamente.
Sindrome di Beckwith-Wiedeman
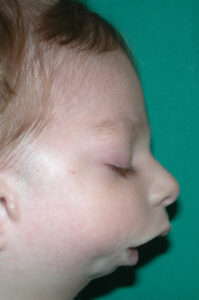
Questa sindrome è la più comune tra le malattia da aumentato accrescimento. La sindrome è dovuta a mutazioni del cromosoma 11 e in corrispondenza del gene che codifica per l’IGF2 (Insulin Growth Factor 2). La funzione di IGF2 mediare dell’azione dell’ormone della crescita. Le alterazioni a carico di questi geni rendono incontrollata la crescita cellulare e provocano le caratteristiche della sindrome.
La sindrome va sospettata se si riscontrano nella madre valori aumentati di AlfaFetoProteina (AFP) e/o BetaHCG e contestualmente all’ecografia morfologica prenatale si riscontrano alcune delle caratteristiche della sindrome (in particolare macrosomia, visceromegalia, anomalie della placenta e polidramniosi). La diagnosi si pone se il bambino presenta 2 criteri maggiori o 1 maggiroe + 2 minori: i criteri maggiori includono la macroglossia, la macrosomia (> 90° percentile), i difetti della parete addominale, l’ipoglicemia neonatale e le anomalie auricolari. La prognosi è sostanzialmente buona e dipende dal tipo e dalla gravità delle malformazioni del soggetto. E’ importante ricordare che questi pazienti fino all’età di circa 10 anni hanno un rischio elevato di sviluppare tumori maligni o benigni (specie nella cavità addominale) e per tale ragione devono essere controllati nel tempo mediante dosaggio dell’alfafetoproteina, della BetaHCG, delle catecolamine urinarie, ed ecografaia addominale, con la tempistica di alcuni schemi di protocollo che sono stati recentemente proposti (vedi: http://www.aibws.org). Nei casi in cui la macroglossia determina difficoltà respiratorie, difficoltà alla deglutizione o alla fonazione e sindrome delle apnee notturne (OSAS) è necessario un intervento chirurgico che riduce la dimensione della lingua.
Craniostenosi
Sono caratterizzate da una precoce ossificazione di una o più suture della scatola cranica e si manifestano con differenti alterazioni della forma del cranio in funzione del tipo e del numero di suture coinvolte. Spesso si manifestano in forma isolata, raramente associate a quadri sindromici. Presentano un aumento della pressione del liquido intracranico che può avere influenza negativa sulla maturazione neurocognitiva del paziente, le probabilità di interferenze aumentano quando è coinvolta più di una sutura cranica. La diagnosi è fatta attraverso l’esame clinico specialistico e confermata dalla TC o dalla RMN.
Nella Trigonocefalia la fronte appare acuiforme centralmente, come la prua di una barca, gli occhi sono più vicini (ipotelorismo orbitarlo) e le aree laterali (temporali) sono appiattite. Si associa spesso a piccoli difetti cardiaci. Non ci sono disturbi sensoriali specifici.Solo le forme più gravi vengono trattate chirurgicamente.
La Plagiocefalia è tra le craniostenosi quella che più coinvolge il volto. Si presenta con sinostosi della sutura emicoronale e del basicranio omolaterale. Troviamo deformità della fronte con appiattimento e retrusione del lato affetto, l’orbita è deforme e più retrusa, lo zigomo è allargato e il naso è deviato controlateralmente configurando nell’insieme una scoliosi facciale che si associa alla deformità cranica. Spesso si associano alterazioni della motilità oculare, della postura della testa e/o dell’intero busto. Con un adeguato trattamento si ottiene la normalizzazione della morfologia craniofacciale, della postura e della funzione oculomotoria, prevenendo la deformità del volto.
Scafocefalia è la più frequente delle craniostenosi (1 caso ogni 3000 nascite), la testa è stretta e allungata,con presenza di “bossing” frontale e/o occipitale, non interessa il volto.
Brachicefalia e Forme complesse, le suture del cranio possono essere interessate in svariate modalità configurando quadri clinici denominati in funzione della deformità che producono.
Dobbiamo infine ricordare che esistono una serie di deformità del cranio non causate da precoce ossificazione delle suture, definite POSIZIONALI, in quanto attribuite alla posizione del feto durante la vita intrauterina. Alcune di queste condizioni possono beneficiare di trattamenti ortopedici con caschi dinamici.
Cranio-facio-stenosi
(Sindrome di Crouzon/Apert/Pfiffer)
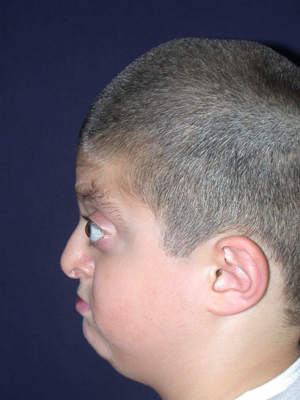
La prima fase chirurgica è spesso la cranioplastica per interrompere le suture chiuse,espandere il cranio e le orbite al fine di ottenere una crescita cranica più armonica evitando l’eccessiva crescita verso l’alto del cranio e conferendo alle orbite un’adeguata profondità al fine di evitare le lussazioni dei bulbi oculari,le ulcere corneali e le alterazioni della visione.
La seconda fase prevede l’avanzamento delle ossa de volto e si esegue quando si manifestano problemi di respirazione o oculari.
I disturbi della respirazione possono manifestarsi già alla nascita ed essere tali da dover utilizzare un’ ossigenazione forzata notturna (C – PAP ) o da richiedere la tracheotomia. Anche le lesioni corneali e le lussazioni dei bulbi oculari possono manifestarsi precocemente.
I difetti di crescita delle ossa craniofacciali possono essere corretti con tecnica di osteodistrazione mediante l’uso di appositi apparecchi.
La displasia frontonasale è un quadro particolare con craniostenosi e ipertelorismo orbitarlo che presenta la sintomatologia delle craniostenosi ma che necessita di una specifica tecnica chirurgica correttiva.
Acondroplasia
Malformazione caratterizzata da un difettoso accrescimento scheletrico per un disturbo dei processi di maturazione e di ossificazione a livello della cartilagine di coniugazione nelle ossa lunghe degli arti: queste rimangono corte, pur accrescendosi normalmente nelle altre direzioni, così come normalmente si accrescono le ossa del cranio, della faccia e del tronco. Il soggetto acondroplasico pertanto manifesta una forma di nanismo detto disarmonico, in quanto caratterizzato da una sproporzione tra le dimensioni degli arti e il resto del corpo. E’ una malattia ereditaria a trasmissione dominante (anche se l’80% dei casi sono sporadici, cioè dovuti a una mutazione genetica insorta ex novo e non ereditata dai genitori). La loro statura in età adulta può variare da 1 a 1,40 m. La testa è piuttosto grossa, il naso largo e appiattito alla radice, il collo corto piantato su un tronco normalmente sviluppato in altezza e che contrasta fortemente con le braccia e le gambe cortissime e tozze. La radiografia dello scheletro mostra le ossa lunghe degli arti spesse e massicce, alle cui estremità però manca quasi completamente la porzione cartilaginea a carico della quale l’osso si sviluppa in lunghezza. L’intelligenza vivace e brillante spesso li aiuta a superare le difficoltà di un’esistenza tra individui “normali”. La terapia è in pratica inesistente la correzione precoce delle deformità e l’allungamento degli arti, attualmente reso possibile dall’applicazione di sofisticate tecniche ortopediche, consente un buon miglioramento delle funzioni motorie e dell’aspetto estetico. Dal punto di vista craniofacciale le forme più lievi di iposviluppo del terzo medio si possono curare con ortopedia/ortodonzia. Le forme più gravi necessitano un avanzamento chirurgico del mascellare.